Example on computing ionization potential and electron affinity from self-consistent GW for Nitrogen molecule
Here we provide a simple example on how to run Green/Weakcoupling for computing ionization potential (IP) for nitrogen molecule (Geometry obtained for GW100 dataset).
Prerequisites: you must have the mbpt compiled and the green-mbtools python package installed.
Change to a new directory where you will keep your simulations, create a directory for the simulation.
Then create a file with the atom positions in the atom.dat:
N 0.0000 0.0000 0.0000
N 0.0000 0.0000 1.0977The remaining parameters will be specified on the command line.
We then obtain input parameters and the initial mean-field solution by running pySCF via the init_data_mol_df.py script:
python <source root>/green-mbpt/python/init_data_mol_df.py \
--atom atom.dat --basis ccpvdz --xc PBE0We use the ccpvdz basis-set and hybrid PBE0 exchange correlation potential as reference. Not specifying the xc would run Hartree-Fock.
The primary goal of this example is to run a molecular example using fully self-consistent GW and perform analytical continuation to obtain photoelectron spectra and IP.
After that we will run the GW approximation
<install dir>/bin/mbpt.exe --scf_type=GW --BETA 1000 \
--grid_file ir/1e5.h5 --itermax 40 --results_file sim.h5 \
--mixing_type CDIIS --diis_start 2 --diis_size 5 --mixing_weight 0.3 \
--jobs SCHere we run the self-consistent GW approximation at inverse temperature , we use an IR nonuniform grid for .
We then store results into sim.h5 file.
Now we will use the green-mbtools package to perform Nevanlinna analytical continuation from the imaginary axis to real axis.
python <source root>/green-mbtools/examples/nvnl_analysis_mol.py --input input.h5 --sim sim.h5 --iter -1 --beta=1000 \
--grid_file ir/1e5.h5 --out ac_out.h5 \
--e_min -5.0 --e_max 5.0 --n_omega 4000 --eta 0.01This will run Nevanlinna analytical continuation for the data obtained at the last iteration. The output will be stored in the group ac_out.h5.
A plot for the specrta can then be obtained with the plot_dos_mol.py script:
python <source root>/green-mbtools/examples/plot_dos_mol.py --ac_out nvnl_out.h5This will read the analytically continued data and plot it to an <output_dir>/spectra.pdf file. In addition, it will also print the ionization potential (IP) and elecrton affinites (EA).
The resultant spectra looks like:
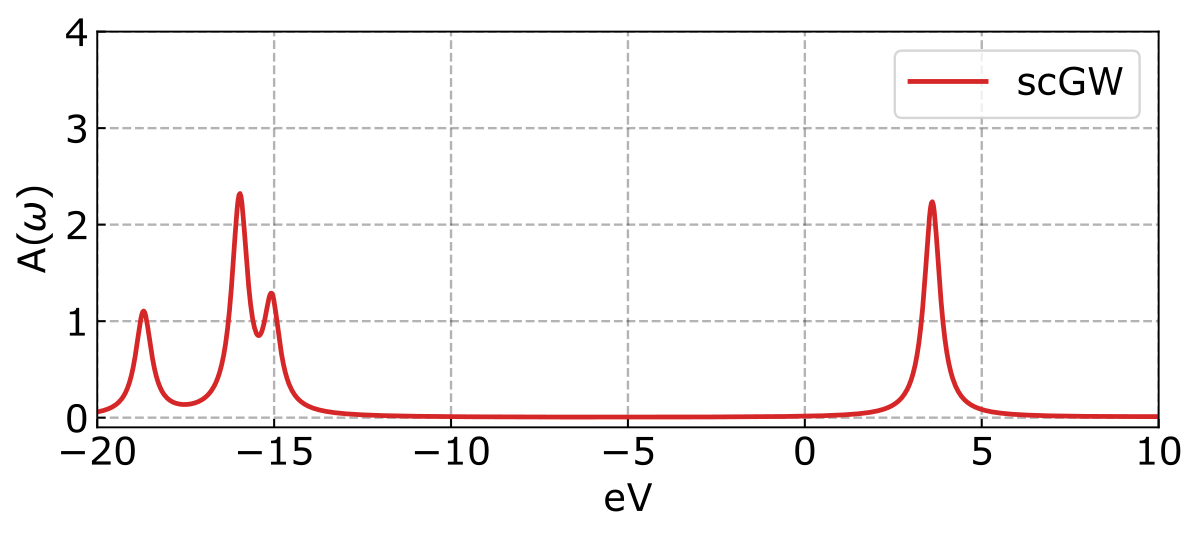
And the ionization potentials (IP) and electron affinities (EA) are displayed as follows.
Lowest 4 Ionization Potentials:
GW IP 1: -15.964 eV
GW IP 2: -15.964 eV
GW IP 3: -15.066 eV
GW IP 4: -18.685 eV
Lowest 4 Electron Affinities:
GW EA 1: 3.604 eV
GW EA 2: 3.604 eV
GW EA 3: 15.143 eV
GW EA 4: 21.321 eVIt is important to highlight that the quasiparticle energies are ordering according to the mean-field reference. As a result, the quasiparticle energies may not be arranged in increasing order because of corrections from dynamic self-energy.