Example to compute GW band structure
Here we provide a simple example on how to run Green/Weakcoupling on the example of a periodic silicon on 6x6x6 lattice. Prerequisites: you must have both the mbpt and the ac executables compiled,
and the green-mbtools python package installed.
Change to a new directory where you will keep your simulations, create a directory for the Si simulation, and create the file a.dat containing the following unit cell information:
0.0, 2.7155, 2.7155
2.7155, 0.0, 2.7155
2.7155, 2.7155, 0.0Then create a file with the atom positions in the atom.dat:
Si 0.0 0.0 0.0
Si 1.35775 1.35775 1.35775The remaining parameters will be specified on the command line.
We then obtain input parameters and the initial mean-field solution by running pySCF via the init_data_df.py script:
python <source root>/green-mbpt/python/init_data_df.py \
--a a.dat --atom atom.dat --nk 6 \
--basis gth-dzvp-molopt-sr --pseudo gth-pbe --xc PBEHere we use the gth-dzvp-molopt-sr basis with the gth-pbe psudopotential and run DFT mean-field approximation with a PBE exchange correlation potential.
The primary goal of this example is to generate the band structure of Silicon. For that, we require the overlap matrix and one-electron Hamiltonian along the high-symmetry path in addition to the density functional solution.
We set the high-symmetry path to WGXWLG to reproduce the results from [Phys. Rev. B 106, 235104].
To calculate these quantities, we run the script init_data_df.py again, but this time with an additional --job sym_path flag:
python <source root>/green-mbpt/python/init_data_df.py \
--a a.dat --atom atom.dat --nk 6 \
--basis gth-dzvp-molopt-sr --pseudo gth-pbe --xc PBE \
--job sym_path --high_symmetry_path WGXWLG --high_symmetry_path_points 100Note that it is possible to run both jobs at once using the flag --job init sym_path.
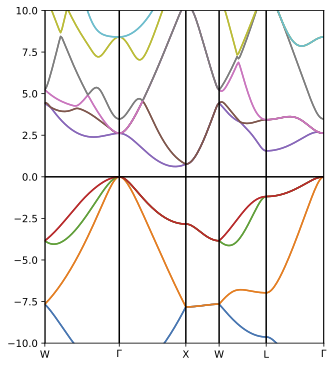
This marks the completion of input data generation and initial mean-field calculation. Before running the GW calculation, it is informative to analyse the mean-field band structure. There are several different ways to obtain the band structure from a converged DFT or HF calculation. Perhaps the fastest approach is to perform k-point interpolation using the green-mbtools python package.
Specifically, we can use the wannier_fock.py for Silicon in the following manner:
python wannier_fock.py --input input.h5 --celltype 'fcc' --bz_type 'fcc' \
--bandpath W G X W L G --bandpts 100The resultant band structure (saved as a PDF file) is expected to look like:
We will now run the GW approximation:
We then store bulk results into Si.h5 file. After the self-consistent simulation is finished band-path interpolation along the high-symmetry path
will be evaluated and stored in Si_hs.h5.
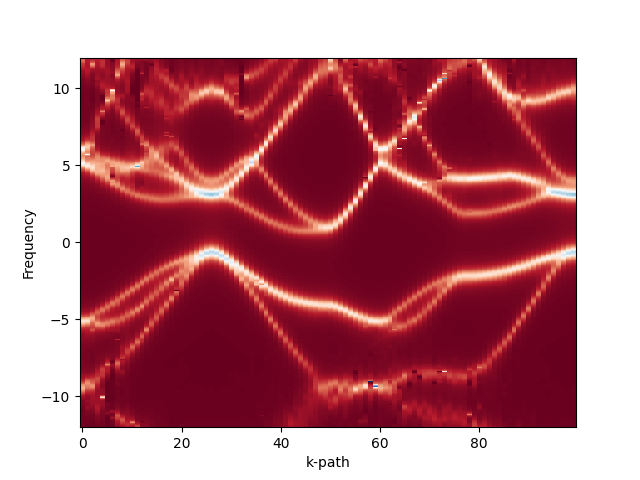
To obtain the spectral function for silicon run
<install dir>/bin/ac.exe --BETA 100 --grid_file ir/1e4.h5 \
--input_file Si_hs.h5 --output_file ac.h5 --group G_tau_hs \
--e_min -5.0 --e_max 5.0 --n_omega 4000 --eta 0.01 \
--kind NevanlinnaThis will run Nevanlinna analytical continuation for the data obtained at the last iteration for all k-points. The output will be stored in the group
G_tau_hs of the output HDF5 file ac.h5.
A plot for the band structure can then be obtained with the plot_bands.py script:
python <source root>/green-mbpt/python/plot_bands.py \
--input_file ac.h5 --output_dir bandsThis will read the analytically continued data and plot it to an <output_dir>/bands.png file. In addition, it will create a plain-text data file for every k-point along the chosen path inside the <output_dir> directory.
The expected band structure plot should look as following